Skip to content
主頁
焦點健聞
健康專題
新冠病毒
三高與中風
糖尿病
肥胖問題
婦科疾病
懷孕與生育
耳鼻喉疾病
兒童疾病
呼吸系統疾病
長者/Young Old 健康
濕疹/皮膚問題
流感
精神健康
痛風
腸胃疾病
認知障礙症 vs 柏金遜
失眠與睡眠質素
近視與眼科疾病
痛症
牙周病/牙齒健康
健康生活
健康減肥
中醫治療
運動與健康
性行為
營養與飲食
癌症360
淋巴癌
肺癌
乳癌
肝癌
大腸癌
卵巢癌
前列腺癌
鼻咽癌
子宮頸癌
胰臟癌
膀胱癌
胃癌
皮膚
Search ...
脊髓肌肉萎縮症
焦點健聞
【罕見病】初生嬰篩查早發現早治療助健康成長 養和港怡已免費篩查逾700嬰
2026年2月3日
編輯推介
脊髓肌肉萎縮症症狀肌肉無力萎縮 新生嬰篩查確診SMA1 接受基因療法終生打一針提升存活率
2023年12月18日
焦點健聞
罕見病藥費分擔原則:不因病致貧 研放寬經濟審查 設資產界線折扣制
2018年10月3日
脊髓肌肉萎縮症
【有片】SMA知多少? 脊髓肌肉萎縮症患者分四型
2018年5月7日
焦點健聞
養和義工隊為「脊髓肌肉萎縮症」籌36萬 患者周佩珊:新藥帶來希望
2018年3月25日
最新文章
甲狀腺癌警號:頸有腫塊、呼吸吞嚥困難 女性患病風險較高
糖尿病導致肌少症?4招趕走肌少症
【罕見病】初生嬰篩查早發現早治療助健康成長 養和港怡已免費篩查逾700嬰
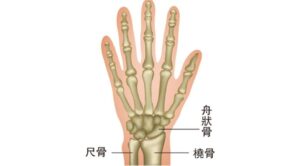
【媽媽手】前臂肌肉繃緊招手腕痛 簡單自我測試風險有多高
外遊遇急病創傷意外 返港治療醫療運送服務流程、風險知多啲
熱門文章
[tptn_list daily=1]
Scroll to Top
Search ...