X
參加明報高中生升學博覽會2025 (7月12-13日),一站式接收本地海外升學資訊,以及聯招放榜前改選秘技
立即報名
Skip to content
主頁
焦點健聞
健康專題
新冠病毒
三高與中風
糖尿病
肥胖問題
婦科疾病
懷孕與生育
耳鼻喉疾病
兒童疾病
呼吸系統疾病
長者/Young Old 健康
濕疹/皮膚問題
流感
精神健康
痛風
腸胃疾病
認知障礙症 vs 柏金遜
失眠與睡眠質素
近視與眼科疾病
痛症
牙周病/牙齒健康
健康生活
健康減肥
中醫治療
運動與健康
性行為
營養與飲食
癌症360
淋巴癌
肺癌
乳癌
肝癌
大腸癌
卵巢癌
前列腺癌
鼻咽癌
子宮頸癌
胰臟癌
膀胱癌
胃癌
皮膚
Search ...
特發性肺纖維化
呼吸系統疾病
間質性肺病成因多 可致乾咳、呼吸困難、肺功能轉差 及早發現藥物控制免惡化
2022年4月27日
焦點健聞
罕見病組織成功游說 肺纖新藥 藥廠允用藥兩年後免費
2018年3月8日
最新文章
疫情後兒童近視率急升眼科醫生指阿托品眼藥水助紓近視加深
睡眠中磨牙不自覺 致晨起面部痠脹、牙齒敏感兼頭痛?或與精神壓力有關
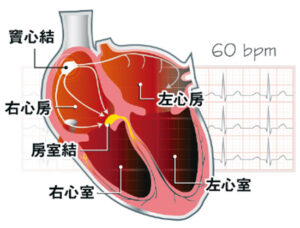
心房顫動:心跳速率力度紊亂致心臟顫抖 中風機率增5倍 了解「脈衝場消融術」
50歲後「準備變老」必備3支柱:身體健康、社交健康和財務健康
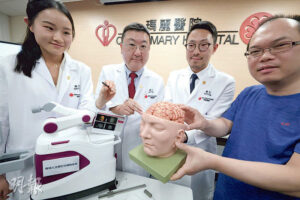
【腦癇症】機械臂微創手術確認癲癇病源 免開顱更精準 手術時間減半
熱門文章
[tptn_list daily=1]
Scroll to Top
Search ...